Affecting more than 100,000 American children and adults, sickle cell disease (SCD) is an inherited hemoglobinopathy that results when a single-nucleotide mutation in the ß-globin gene yields an abnormal “sickle” hemoglobin (HbS). In a low oxygenation state commonly triggered by infection, dehydration, stress or extreme temperatures, HbS forms rigid polymers that cause normally biconcave red blood cells (RBCs) to become elongated, rigid and curved on the ends, resembling a sickle shape.
These stiff, severely deformed sickle RBCs tend to stick together and block or restrict blood flow. Because sickle RBCs are prone to hemolysis, they have a far shorter circulating lifespan than normal RBCs, resulting in chronic anemia.
Most severely affected are individuals with the HbSS (sickle cell anemia) or HbS-ß0-thalassemia genotypes, in whom high levels of HbS and hypoxic trigger events lead to recurrent bouts of severely painful vaso-occlusive crises (VOCs). Acute chest syndrome, severely painful joints, splenic sequestration and stroke head a long list of common serious acute and chronic complications of severe SCD (Table).
Expanding SCD Treatment Options
Two mainstay treatments have been used for many years in SCD patients in an effort to limit VOCs and reduce other serious complications. The first is RBC transfusion to acutely or chronically reduce the level of circulating HbS and improve oxygen delivery. The other is hydroxyurea, potent oral antimetabolite used in higher doses to treat certain cancers. Through an incompletely understood means, hydroxyurea induces production of fetal hemoglobin (HbF), which inhibits intracellular HbS polymerization and the sickling phenomenon. In adults, hydroxyurea can substantially reduce the frequency of severe painful vaso-occlusive episodes, acute chest syndrome, hospitalizations and blood transfusion requirements.1 In children with SCD ages 2 years and older, hydroxyurea has been shown to decrease the frequency of pain crisis and childhood hospitalization.2
But the limited benefits of hydroxyurea and transfusion to reduce VOCs and other SCD complications have spurred development of newer treatments. By blocking a protein that causes stickiness between vascular endothelial cells, RBCs, platelets and leukocytes, the humanized monoclonal antibody crizanlizumab (ADAKVEO) has been shown to further reduce VOC incidence in subjects both treated and not treated with hydroxyurea. The U.S. Food and Drug Administration (FDA) approved ADAKVEO in 2017 after a pivotal trial showed that addition of the drug achieved a 45 percent reduction in the median annual rate of VOCs, from 2.98 to 1.63 events.3
Supplementation of hydroxyurea with twice-daily oral L-glutamine (ENDARI), also approved by FDA in 2017, was separately proven to reduce the median number of VOCs by about one-quarter in a placebo-controlled study in 230 adults and children with sickle cell anemia or HbS-ß0-thalassemia. ENDARI therapy also resulted in fewer hospitalizations due to sickle cell pain and a lower incidence of acute chest syndrome.4,5
Two years later in 2019, FDA approved voxelotor (OXBRYTA), another oral medication that, by increasing hemoglobin affinity for oxygen, inhibits HbS polymerization, thereby reducing RBC sickling and hemolysis-associated anemia.6
These newer treatment options have proven utility in reducing VOCs and other comorbidities, but they are far from a cure, particularly for more severely affected children and adults with more frequent VOCs. According to the U.S. Centers for Disease Control and Prevention (CDC), the average life expectancy for persons with SCD is still shortened by more than 20 years compared with the general population; when factoring in quality of life, more than 30 years are lost.7 So despite the availability of several drug treatment options that didn’t exist a decade ago, many patients with SCD continue to suffer from severe morbidity and shortened lifespan.
Limitations of Donor Stem Cell Transplantation
In children and qualifying young adults with SCD who have the HbSS or HbS-ß0-thalassemia genotypes and one or more specific manifestations of severe disease,* allogeneic hematopoietic stem cell transplantation (HSCT) is curative in more than 90 percent of cases.8 But outside of investigational clinical trials, HSCT is appropriate only for SCD patients who have a fully HLA-matched sibling without SCD in order to avoid major risks of potentially life-threatening graft-versus-host disease (GVHD).
*Stroke, recurrent acute chest syndrome, recurrent severe crisis pain, recurrent priapism, impaired neuropsychological function with evidence of cerebral infarction, sickle cell nephropathy, stage I or II sickle lung disease, bilateral proliferative retinopathy and major visual impairment, osteonecrosis of multiple joints, and red cell alloimmunization with more than two antibodies during long-term transfusion therapy. [Walters MC, De Castro LM, Sullivan KM, et al. Indications and results of HLA-identical sibling hematopoietic cell transplantation for sickle cell disease. Biol Blood Marrow Transplant 2016 Feb;22(2):207-11].
Unfortunately, fewer than one in five children with SCD have an HLA-matched sibling, leaving all others without access to HSCT. And even in the optimal circumstance where the donor is an HLA-matched sibling, allogeneic HSCT has real risks, including:
• A roughly 15 percent overall risk of developing acute or chronic GVHD, increasing with transplant age;9
• A risk of future infertility resulting from myeloablative conditioning;10,11 and
• A risk of premature death (an overall seven percent probability of five-year mortality, increasing about 10 percent with each one-year increase in transplant age).9
Fortunately, a second curative strategy for SCD has finally arrived: gene therapy. And because it involves use of autologous hematopoietic stem cells (HSCs), this novel treatment approach isn’t burdened by the HSCT-associated risks of serious or potentially life-threatening GVHD, nor restricted to a small subpopulation of SCD patients fortunate enough to have an HLA-matched full sibling.
Two SCD Gene Therapies, Two Approaches
Conventional gene therapies exert their therapeutic effect through insertion of a gene encoding a missing protein that plays a critical role in an enzymatic or metabolic pathway. Novartis’ ZOLGENSMA, for example, is an adeno-associated viral (AAV) vector-based gene therapy that stops the progression of spinal muscular atrophy by replacing the missing or nonworking human survival motor neuron (SMN1) gene. BioMarin Pharmaceuticals’ ROCTAVIAN and CSL Behring’s HEMGENIX similarly use an AAV vector to deliver genes respectively encoding functional coagulation factors VIII and IX missing in persons with hemophilia A and B.
In December 2023, the FDA approved not one but two novel, single-treatment gene therapies for the treatment of SCD patients age 12 years and older with a history of VOCs. One of these two therapies (LYFGENIA) applies the classical functional protein replacement model, while the other (CASGEVY) employs an entirely novel approach to overcoming HbS-mediated RBC pathophysiology.
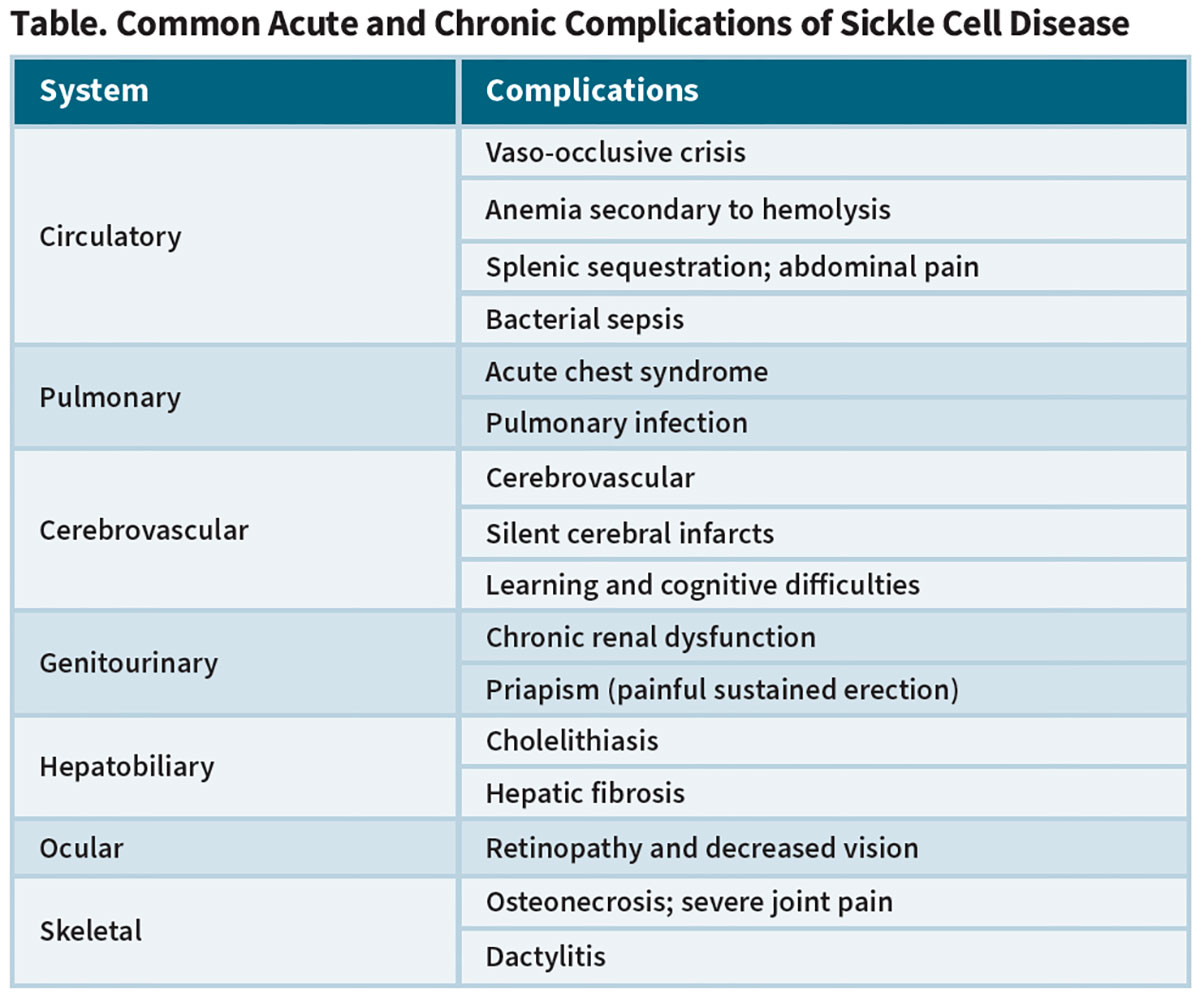
LYFGENIA (lovotibeglogene autotemcel) (bluebird bio). Similarly to the way that approved hemophilia gene therapies work through insertion of genes encoding functional factor VIII and IX, the LYFGENIA procedure employs a recombinant replication-defective lentiviral vector (LVV) to introduce copies of a modified ß-globin gene (ßA-T87Q) into the genome of the patient’s bone marrow-harvested autologous CD34+ cell-enriched HSCs (Figure 1). ßA-T87Q combines with native ß-globin to yield an anti-sickling hemoglobin variant (HbAT87Q) whose oxygen-binding affinity and dissociation properties are similar to normal hemoglobin. And because this generated HbAT87Q both reduces intracellular and total HbS levels and acts to inhibit polymerization of HbS, it also limits the sickling of RBCs.
A pair of clinical trials that treated a total of 47 SCD patients with a history of recurrent VOCs or stroke through up to five years of follow-up (median of 35.5 months) generated these key results:12,13
• All 47 patients exhibited strong, stable production of the anti-sickling hemoglobin variant, with a median HbAT87Q exceeding 40 percent.
• Median total hemoglobin increased from 8.7 g/dL at pre-treatment baseline to 11.8 g/dL at the last visit.
• Of 34 evaluable patients, 32 (94 percent) had complete resolution of severe VOCs; 30 had complete resolution of all VOCs; this compares to a median of 3.0 severe VOCs and 3.5 total VOCs per year in the two years prior to LYFGENIA treatment.
• Patients reported sustained improvements in pain intensity, pain interference and fatigue, up to 48 months of follow-up.
No evidence of graft failure, replication-competent lentivirus or vector-mediated oncogenesis was observed in any patient enrolled in the LYFGENIA clinical development program, extending back more than eight years.
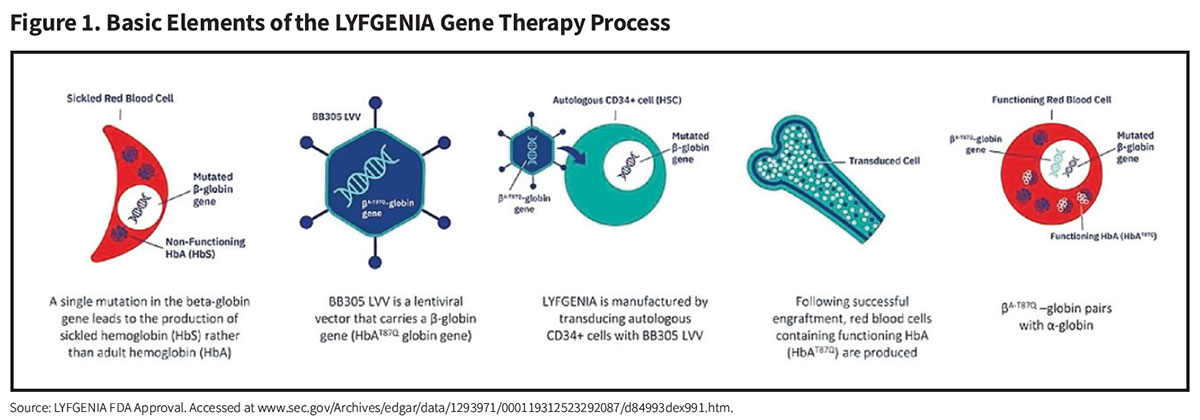
CASGEVY (exagamglogene autotemcel) (Vertex Pharmaceuticals). While LYFGENIA reduces RBC sickling by inserting functional copies of a modified functional ß-globin gene, the CASGEVY procedure instead employs an entirely new nonviral gene editing strategy to reactivate dormant fetal hemoglobin (HbF) production and drive down HbS levels (Figure 2).
Harvested autologous CD34+ cell-enriched HSCs are collected from the patient and edited by a technology called “clustered regularly interspaced short palindromic repeats” (CRISPR). Sometimes referred to as “genetic scissors,” CRISPR directs an enzyme called Cas9 to a specific region of a gene called BCL11A, whose function is to shut off the production of the fetal form of human hemoglobin (HbF) following birth. CRISPR-Cas9 deactivates BCL11A by cutting its DNA, resulting in reactivated HbF production by these gene-edited HSCs.
In June of this year, Vertex reported results from CLIMB SCD-121, a 24-month Phase III trial of CASGEVY in patients ages 12 to 35 years with a history of ≥2 VOCs per year. Here are key findings in 46 SDC patients who received CASGEVY, including 17 patients who completed at least two years of follow-up:14
• Twenty-nine of 31 (93.5 percent) evaluable patients were free of severe VOCs for ≥12 consecutive months (VF12); all 31 patients (100 percent) were free from hospitalizations for VOCs for ≥12 consecutive months.
• In patients achieving VF12, the mean VOC-free duration was 25.4 months (range 18.0-48.7 months).
• For all treated patients, the mean total hemoglobin was 11.9 g/dL at month 3 and ≥11 g/dL from month 6 onward.
• The mean HbF as a percentage of total hemoglobin was 37.1 percent at month 3 and generally ≥40.0% from month 6 onward with pancellular distribution (≥95 percent of RBCs express HbF).
• Patients reported sustained and clinically meaningful improvements in their health-related quality of life, across physical, emotional, social/family and functional well-being parameters, pain experience and overall health status.
• No patients had serious adverse events considered related to CASGEVY therapy.
While CRISPR technology is being investigated for a variety of therapeutic applications, CASGEVY has the distinction of being the first-ever CRISPR-based gene editing therapy to be approved in the U.S.
A second one-time CRISPR-based autologous gene editing therapy, Editas Medicine’s renizgamglogene autogedtemcel (“reni-cel”), is also in late-stage clinical development for treatment of SCD. Like CASGEVY, reni-cel introduces edits that reactivate production of HbF.
While just two of 18 adult severe SCD patients dosed with reni-cel have been monitored for more than a year, none of these 18 patients have experienced a VOC over a mean of six months. Following reni-cel infusion, total hemoglobin levels rapidly normalized, with a sustained HbF level exceeding 40 percent through the last follow-up.15 Massachusetts-based Editas plans to enroll a total of 45 severe SCD patients to evaluate reni-cel’s efficacy, safety and tolerability.
Implementing SCD Gene Therapy
Both the CASGEVY and LYFGENIA therapy procedures require providers with specialized experience in stem cell transplantation. Individualized decisions must be made at each treatment phase, including mobilization of autologous HSCs, myeloablative drug conditioning to create space for the gene-modified HSCs to engraft in the bone marrow, use of RBC transfusions to reduce the risk of SCD-related complications, and management of adverse events that may follow infusion of gene-modified HSCs.
Vertex and bluebird bio are each engaging with carefully selected hospitals to qualify them for inclusion in their participating treatment center networks. To date, bluebird bio has activated — or is in the process of activating — at least 55 “Qualified Treatment Centers,” including 25 children’s hospitals. Vertex is on track to activate a similar number of “Authorized Treatment Centers.”
Like other gene therapies licensed to date, CASGEVY and LYFGENIA are expensive, with list prices of $2.2 million and $3.1 million, respectively. However, less than a week after FDA approval, bluebird bio disclosed that it had reached an outcomes-based agreement with a third party payer organization representing approximately 100 million covered lives, and was in discussions with other large commercial payers and more than 15 state Medicaid agencies representing 80 percent of individuals with SCD in the U.S.16 Under its announced Cell and Gene Therapy Access Model, beginning next year the U.S. government’s Centers for Medicare and Medicaid Services (CMS) similarly plans to negotiate outcomes-based agreements with the two manufacturers on behalf of participating state Medicaid agencies; between 50 percent and 60 percent of SCD patients are covered by the Medicaid program, according to CMS.17
Modeling CASGEVY and LYFGENIA against the current standard of care, the Institute for Clinical and Economic Review (ICER) calculated a Health Benefit Price Benchmark ranging between $1.35 million and $2.05 million. More importantly, ICER concluded that, while acknowledging uncertainties about long-term outcomes, “both CASGEVY and LYFGENIA are likely to substantially improve quality and length of life among patients with SCD.”18
Vertex estimates that some 16,000 U.S. SCD patients may be eligible for a one-time gene therapy that offers the potential of a durable functional cure by eliminating severe VOCs and associated hospitalizations.19 While the evidence to date is highly promising, it remains to be seen whether a single infusion of genetically modified HSCs can actually provide a long-term cure. In the meantime, it is up to providers, insurers and government policymakers to work together to assure every qualifying individual with severe SCD has access to these therapies and the opportunity to live a long, healthy life.